Identifiée pour la première fois, en 1969, par le neurologue français Marie Joubert, ce trouble génétique rare, affecte les garçons et les filles. Le syndrome de Joubert, également appelé syndrome cérébello-oculo-renal, est parfois détecté dès la naissance ou un peu plus tard.
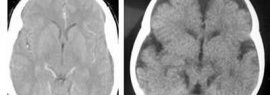
Le syndrome de Joubert est une maladie d'origine génétique, résultant d'une série d'anomalies dans au moins 5 gènes situés sur divers chromosomes, notamment les chromosomes 2, 6, 9, 11 et 12. Ces gènes défectueux sont responsables du développement incomplet du vermis cérébelleux et du tronc cérébral, au cours de la grossesse.
Symptômes du syndrome de Joubert
Le tableau clinique du syndrome de Joubert est lié, d'une part, à la malformation du cervelet, notamment au sous-développement du vermis cérébelleux qui est responsable de l'équilibre et de la coordination musculaire, et d'autre part, aux anomalies du tronc cérébral.
Les manifestations sont perceptibles chez le nourrisson avec un retard du développement moteur, des anomalies respiratoires ou des troubles du rythme respiratoire, des mouvements anormaux des yeux et un faible tonus musculaire (hypotonie). Au cours de la petite enfance, les difficultés de coordination des mouvements musculaires volontaires (ataxie), les troubles des mouvements oculaires (apraxie oculomotrice) sont observables.
Ces manifestations sont variables d'un enfant à un autre (de léger à sévère) avec notamment une déficience intellectuelle qui peut légère ou modérée, des difficultés d'apprentissage, des difficultés au niveau de la motricité manuelle, un faciès dysmorphique marqué par un front proéminent, des paupières tombantes (ptosis), un nez retroussé.
Les atteintes hépatiques ou rénales, une fente labiale, des doigts et des orteils complémentaires (polydactylie) peuvent être présents chez certains enfants. Chez d'autres, les symptômes peuvent être caractérisés par des atteintes rétiniennes, des fentes palatines, des malformations cardiaques ou un dysfonctionnement hormonal.
Traitement du syndrome de Joubert
Le syndrome de Joubert est une anomalie congénitale qui ne peut pas être guérie, car il n'existe aucun remède. Cependant, le traitement qui est essentiellement pluridisciplinaire et symptomatique peut grandement contribuer à l'amélioration de la qualité de vie des malades.
Cette prise en charge vise à réduire la gravité des symptômes et à garantir des soins adaptés aux besoins médicaux des patients en vue de les aider à atteindre un potentiel optimal sur le plan physique et intellectuel.
Pour ce faire, l'approche thérapeutique multidisciplinaire peut nécessiter l'expertise d'un ophtalmologue en vue de traiter les manifestations oculaires ; l'intervention du kinésithérapeute est recommandée lorsque des mesures rééducatives sont nécessaires en présence d'une hypotonie musculaire et d'un retard du développement moteur.
Les manifestations neurologiques et les atteintes rénales peuvent être traitées respectivement par les neurologues et les néphrologues. Les fentes palatines peuvent être corrigées par le biais d'une chirurgie. L'adaptation de la prise en charge requiert également des bilans neurophysiologiques afin de mieux appréhender l'évolution des capacités cognitives. En plus du suivi médical, le patient et sa famille doivent être soutenus psychologiquement.